Creating Datasets¶
Overview¶
Datasets are like documents in a word processor. To start working with data on SolveBio, you'll need to create a new, empty dataset. Datasets have both a schema (dataset fields) and contents (dataset records). On SolveBio, unlike other database systems, you don't need to know your schema in advance (fields are automatically detected from imported data). However, in many cases crafting a schema (i.e. setting dataset fields) can help avoid issues with data types and field names.
Create a Dataset¶
To create a dataset, supply a full path in the following format: <domain>:<vault>:<path> (e.g. myDomain:MyVault:/folder/with/dataset or ~/folder/with/dataset to use your personal vault). The path must be within a vault where you have write-level access (such as your personal vault).
1 2 3 4 | from solvebio import Dataset # Create a new, empty dataset in your personal vault (represented by "~/") dataset = Dataset.get_or_create_by_full_path('~/my_dataset') |
1 2 3 4 5 6 7 8 | library(solvebio) # Specify where you want the new dataset vault <- Vault.get_personal_vault() dataset_full_path <- paste(vault$full_path, "/r_examples/my_dataset", sep=":") # Create a new, empty dataset dataset <- Dataset.get_or_create_by_full_path(dataset_full_path) |
When creating the dataset you can supply a number of optional parameters:
description: A description (text) of the dataset.fields: A list of field objects (see below).capacity: A performance optimization for datasets that will have tens or hundreds of millions of records. Default is "small" but can be set to "medium" or "large". This cannot be changed once it has been set.metadata: A dictionary of key/value pairs which can be associated to the dataset.tags: A list of strings (tags) that can be associated to the dataset.
Once the dataset is created it will be empty (containing no records) and consist of the default SolveBio _id field and any other fields that may have been added by the fields parameter.
Dataset Fields¶
By default, new fields are automatically detected by the import system. You can also provide a list of fields (i.e. a template) using the fields parameter. This lets you explicitly set field names and titles, data types, ordering, descriptions, and entity types for each field. See dataset field reference for more info.
The following example creates a new dataset using a template with two fields:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 | from solvebio import Dataset from solvebio import DatasetField fields = [ { "name": "my_string_field", "description": "Just a string", "data_type": "string", "is_list": False, "is_hidden": False, "ordering": 0 }, { "name": "gene_symbol", "description": "HUGO gene symbol", "data_type": "string", "entity_type": "gene" } ] dataset_full_path = '~/python_examples/my_fields_dataset' # Fields, capacity, and other optional parameters can be set during dataset creation dataset = Dataset.get_or_create_by_full_path( dataset_full_path, fields=fields, capacity='small' ) # If the dataset already exists, you can add additional fields: DatasetField.create( dataset_id=dataset.id, name="my_new_field", data_type="string") # Fields can also be edited field = dataset.fields("my_string_field") field.description = "A new description" field.save() |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 | library(solvebio) fields <- list( list( name="my_string_field", description="Just a string", data_type="string", is_list=FALSE, is_hidden=FALSE, ordering=0 ), list( name="gene_symbol", description="HUGO Gene Symbol", data_type="string", entity_type="gene" ) ) vault <- Vault.get_personal_vault() dataset_full_path <- paste(vault$full_path, "/r_examples/my_fields_dataset", sep=":") # Fields, capacity, and other optional parameters can be set during dataset creation dataset <- Dataset.get_or_create_by_full_path(dataset_full_path, fields=fields, capacity="small") # If the dataset already exists, you can add additional fields: DatasetField.create( dataset_id=dataset$id, name="my_new_field", data_type="string") |
Field Properties¶
Dataset fields have the following properties:
| Property | Value | Description |
|---|---|---|
| name (required) | string | The "low-level" field name, used in JSON formatted records. |
| data_type | string | A valid data type. |
| description | string | Free text that describes the contents of the field. |
| entity_type | string | A valid SolveBio entity type. |
| expression | string | A valid SolveBio expression. |
| is_hidden | boolean | Set to True if the field should be excluded by default from the UI. Default is False. |
| is_list | boolean | Set to True if multiple values are stored as a list. Default is False. |
| ordering | integer | The order in which this column appears in the UI and in tabular exports. |
| title | string | The field's display name, shown in the UI and in tabular exports. Default is set automatically from the name. |
| is_transient | boolean | Set to True if the field is a temporary field used for the purposes of easier data & expression manipulation during imports & migrations. Default is False. See example for usage. |
| depends_on | list of strings | List of fields that must have expressions run first before this field's expression is evaluated. In other words, what other fields that this field depends on. Default is an empty list. |
| url_template | string | A URL template with one or more "{value}" sections that will be interpolated with the field value and displayed as a link in the dataset table |
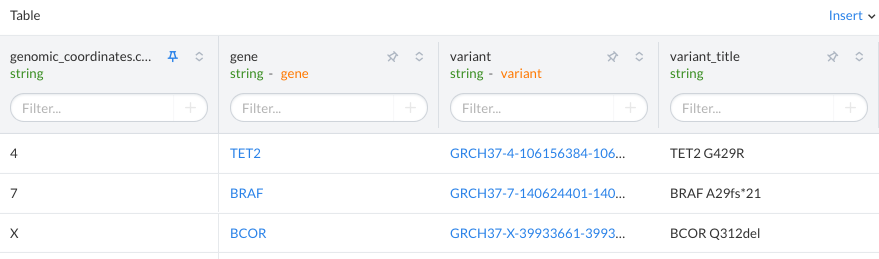
Adding Links to the dataset table
If you add a url_template value to the dataset field, the dataset table will show the value as a link in the SolveBio UI. This is useful for linking out to other sources/websites. The dataset below has links for the gene and variant pages on SolveBio
Modifying Fields¶
Fields can be modified via the API or via the SolveBio UI from several places. Look for the pencil icon next to a dataset field on the dataset's About page or any of the filtering or facets panels.
Data types and field names cannot be changed. Once a field is added to a dataset (either manually or as a result of an import), the field's data type (data_type) cannot be altered in-place. To change the data type of a field, you can perform a dataset migration.
The title, description, url_template, ordering, entity_type can be modified at any time.
Dataset Caveats¶
Reserved Fields¶
SolveBio never alters input data, except in the case of reserved fields. Fields beginning with an underscore such as _id and _commit are considered reserved and may be modified during an import.
The _commit field is always reset during the commit process, and makes it possible to track and log all changes made to a Dataset. The _id field represents the unique ID for each record, which can be used to edit or delete individual records. The value of _id cannot be edited once a record is saved.
Removing Fields¶
Fields cannot be removed in-place.
Fields can easily be hidden to end-users either programmatically or using SolveBio's web interface, but this does not remove the underlying data. To remove a field from a dataset, it must be cloned to a new dataset that does not have the field you intend to delete.
Renaming Fields¶
Field names cannot be renamed, only titles.
While field titles can change (as they are for display purposes only), field names are static and cannot be changed. To change the name of a field, use a dataset migration to create a new field with the desired name.
The following example renames the field of the datasets:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | import solvebio as sb dataset = sb.Dataset.get_or_create_by_full_path("dataset_id") # We want to transform the data by passing target_fields. # Because there is no "rename" functionality, the workaround is to create new fields from with the values from the old fields. Set is_transient=TRUE for the old fields so that they are just temporarily used during data transform, and not included in the output. # The new fields just take the value of the old fields. target_fields = [ { "name": "old_field_name", "is_transient": True }, { "name": "new_field_name", "data_type": 'string', "expression": "record.old_field_name" } ] # Create migration migration = dataset.migrate(target=dataset, target_fields=target_fields) |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 | # Retrieve the source dataset source_dataset <- Dataset.get_by_full_path('solvebio:public:/ClinVar/3.7.4-2017-01-30/Variants-GRCh37') # Create your new target dataset vault <- Vault.get_personal_vault() dataset_full_path <- paste(vault$full_path, "/r_examples/clinvar_renamed", sep=":") target_dataset <- Dataset.get_or_create_by_full_path(dataset_full_path) # We only want data from these fields fields <- list('gene_symbol', 'clinical_significance', 'review_status') # We want to transform the data by passing target_fields. # Because there is no "rename" functionality, the workaround is to create new fields from with the values from the old fields. Set is_transient=TRUE for the old fields so that they are just temporarily used during data transform, and not included in the output. # The new fields just take the value of the old fields. target_fields <- list( list( name='gene_symbol', is_transient=TRUE ), list( name='clinical_significance', is_transient=TRUE ), list( name='review_status', is_transient=TRUE ), list( name='gene', data_type='string', expression='record.gene_symbol' ), list( name='clin_sig', data_type='string', expression='record.clinical_significance' ), list( name='rev_stat', data_type='string', expression='record.review_status' ) ) # Create migration # source_params # fields kwarg returns only the fields defined # limit kwarg pulls only 10 records, remove for all records # target_fields # defines the field transform template migration <- DatasetMigration.create( source_id=source_dataset$id, target_id=target_dataset$id, source_params=list( fields=fields, limit=10 ), target_fields=target_fields ) |
Dataset Capacity¶
The "capacity" of the dataset determines its import, migration and query performance. For most datasets, a "small" capacity will be sufficient. There are three available capacities:
- small - the default value and one that is good for most datasets up to 1 million records.
- medium - for medium sized datasets up to 100 million records.
- large - for large datasets that will have more than 100 million records.
The record limit is not a "hard" limit, e.g. a small dataset could have millions of records, it will just be less performant as more records are added.
Once a dataset is created, its capacity cannot be changed. However, you can always copy the data into a new higher capacity dataset.